Some of the forces that shape biopharma cluster development are constants year after year, such as the emergence of startups from university and research institute labs to develop new treatments, thanks to ideas backed by the brains of researchers and executives, and the bucks of serial entrepreneurs and other investors.
But in recent years, several additional unique circumstances have come to reshape how much and especially where biopharmas choose to grow, Matthew Gardner, CBRE Americas Life Sciences Leader, shared with GEN recently.
One is increased acquisition of lab and manufacturing properties by “mid-cap” biopharmas ranging between $2 billion and $10 billion in market capitalization (share price times the number of outstanding shares), as they seek to better control their supply chains by maintaining their own infrastructure in evolving from research- to commercialization-focused drug developers.
“They might have been more likely to lease in a different circumstance. They’ve definitely caught an opportunity to jump in and take ownership. That has been an ongoing trend, and that has been true coast-to-coast in most of the major centers,” Gardner said.
Among investor-owners, Gardner said, another transition has begun from pure-play biopharma real estate landlords to investors with broader portfolios encompassing healthcare—a reflection of how the two fields are increasingly converging. During December 2025 and January 2026, for example, the public real estate investment trust (REIT) Healthpeak shelled out $600 million to close on the acquisition of a 1.4-million square foot, 29-acre campus on Gateway Boulevard in South San Francisco, CA, from the nation’s largest biopharma REIT, Alexandria Real Estate Equities and BXP (formerly Boston Properties).
Those and other investors aim to cash in on the improving climate for biopharmas seeking to raise capital, from a recovering venture capital market to increased merger-and-acquisition (M&A) activity, and, in recent weeks, a revived market for initial public offerings (IPO).
Another key factor in recent cluster-building cited by Gardner is the “reshoring” of manufacturing in the U.S. by global biopharma giants, whether to satisfy growing demand for treatments—especially obesity drugs—or avoid tariffs, or both. While many of those new facilities are in manufacturing-heavy clusters like North Carolina and Greater Philadelphia, others have spread into Maryland and Virginia (the BioHealth Capital Region), and several new biomanufacturing sites have been built or are under construction in emerging clusters outside the Top 10—a trend GEN plans to explore in the coming weeks.
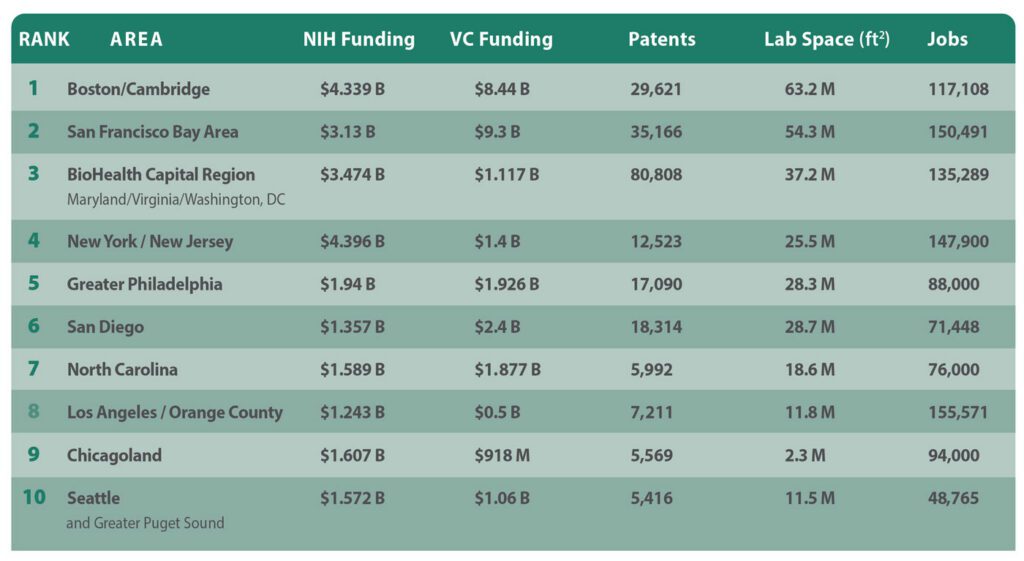
Speaking of top 10 clusters, GEN presents its latest edition of its nationally- and regionally-cited annual A-List of its top 10 U.S. biopharma cluster rankings, designed to show which regions are most competitive in attracting life sciences leaders, companies, and institutions. Over more than a decade, GEN has based its rankings on five criteria:
- Patents: Figures from the Patent Public Search database of the U.S. Patent and Trademark Office, showing the number of patent families containing the word “biotechnology” and towns and cities within a given region or state.
- NIH funding: Figures for NIH funding were taken from the publicly available NIH Research Portfolio Online Reporting Tools (RePORT) database for the current federal fiscal year through May 4, plus all of fiscal year 2025 (October 1, 2024, through September 30, 2025).
- Venture capital funding: Figures for all of 2025 and the first quarter of 2026 as compiled by regional life sciences groups and PitchBook, which joins with the National Venture Capital Association to publish the quarterly Venture Monitor reports.
- Laboratory space: The total-size-of-market figure, in millions of square feet, as furnished by regional life sciences groups. In regions that did not compile such information, the figure cited is the highest by any of several commercial real estate companies, including CBRE Group, Colliers, Cushman & Wakefield, JLL, and Newmark.
- Number of jobs: The preferred sources for job figures were regional life sciences groups. Alternative sources included commercial real estate firms.
1. Boston/Cambridge, MA

Years of growing into the nation’s top biopharma cluster have taken a toll on Boston and adjacent Cambridge, MA: The Wall Street Journal in December highlighted the inability of Boston-area PhDs to find work, while the region faces a glut of life sciences space as biopharmas and real estate developers scale back earlier plans—a 32.7% availability rate according to CBRE, up 70 basis points from Q1 2025. Takeda Pharmaceutical in March eliminated 247 jobs in Massachusetts, where the company has facilities in Lexington, MA, and Cambridge, part of a $1.3 billion restructuring that cut 634 jobs nationwide. Replimune in April chopped 223 jobs at its Woburn, MA, HQ, and Framingham, MA, manufacturing site after the FDA rejected its BLA seeking approval for RP1 [plus Bristol Myers Squibb’s Opdivo® (nivolumab)] for advanced melanoma. In February, Takeda placed 449,140 square feet within three Cambridge buildings on the sublease market, a week after Alexandria Real Estate Equities scrapped plans to convert 401 Park Drive in Boston’s Fenway section into lab space, with CEO and chief investment officer Peter M. Moglia saying the real estate investment trust was pivoting to meet growing demand for office space.
Among the region’s growing life-science companies: Genentech agreed to more than triple its space, growing from 30,000 to 100,000 square feet within One Milestone Street at the Harvard University-owned, Tishman Speyer-developed Enterprise Research Campus in Boston’s Allston section. Hemab Therapeutics (based in Cambridge and Copenhagen) and Seaport Therapeutics (Boston) both priced IPOs on April 30, raising $301.5 million and $254.88 million, respectively—a day after Avalyn Pharma (Boston) garnered $300 million in its IPO. In March, Terrestrial Bio became the first life-science tenant at Allston Labworks (250 Western Avenue) by leasing 42,000 square feet at the mixed-use building within Boston’s Allston neighborhood, while AI Proteins in January inked a 40,000-square-foot lease at 660 Commonwealth Avenue, within Related Beal’s One Kenmore Square in Boston. Regional companies finding buyers in April include Boston-based Kelonia Therapeutics and Cambridge-based Ajax Therapeutics, both to be acquired by Eli Lilly (for up to $7 billion and up to $2.3 billion, respectively) and Framingham-based KalVista Pharmaceuticals, to be acquired by Italy’s Chiesi Group for about $1.9 billion.
Boston/Cambridge enjoys the nation’s largest portfolio of lab space (63.2 million square feet according to industry group MassBio), but was bested by the San Francisco Bay Area in NIH funding (7,037 awards totaling $4.339 billion) following a year of government funding cuts. The region also placed second in VC ($6.85 billion in 2025, says MassBio; $1.59 billion in Q1 2026, according to PitchBook data cited by MassBio), but landed third in patents (29,621 families) and just fifth in jobs (117,108, according to MassBio).
2. San Francisco Bay Area

Santa Clara, CA-based Nvidia and Eli Lilly electrified the annual J.P. Morgan Healthcare Conference, held in downtown San Francisco each January, by announcing a five-year, $1-billion collaboration to create a “Co-Innovation AI Lab” in the region to address key challenges in artificial intelligence (AI) drug discovery, powered by a supercomputer that went live in February. That welcome news aside, more than one-third of the region’s life-science space is available for lease (33.7% as of Q1, according to CBRE). And more space has entered the market: Pfizer confirmed plans in April to shut down its 164,000-square-foot research facility at 181 Oyster Point Blvd. in South San Francisco, CA, shifting employees to remote jobs. Cushman & Wakefield is marketing the space for sublease. Also, on the market in “South City” is a 21,552-square-foot lab building and surrounding 3.65 acres previously occupied by the U.S. Department of Agriculture, which is selling the building for just under $48 million. In May, Foster City, CA-based Gilead Sciences disclosed plans to lay off 108 employees based in Redwood City, CA, (and 84 in Rockville, MD) following its $7.8-billion acquisition of Arcellx.
Not all the recent news is bad: Gladstone Institutes plans early next year to open approximately 20 new labs employing about 300 scientists within the 105,000 square feet it agreed to lease in March at 1450 Owens Street, within Alexandria Real Estate Equities’ Alexandria Center® for Science and Technology–Mission Bay Megacampus. Natera inked a 62,969-square-foot lease at Brittan West in San Carlos, CA, in February. And last fall, Elon Musk’s Neuralink leased the entire approximately 144,000-square-foot 499 Forbes Boulevard in South San Francisco. On the financing side, SF-based Breakout Ventures in March closed its $114-million Fund III, which aims to invest in founder-led companies applying AI in biopharma, while Palo Alto, CA-based Surf Bio, whose lead investor for its only institutional round was Breakout, was acquired by San Diego-based Halozyme Therapeutics for up to $400 million, in a deal announced in January.
San Francisco and its suburbs topped Boston/Cambridge in VC ($7.8 billion in 2025, $1.5 billion in Q1 2026, both according to PitchBook). The Bay Area is second in three criteria: patents (35,166 families), lab space (54.3 million square feet according to Colliers), and jobs (150,491 according to BIOCOM California, but “more than 147,000” according to CBRE, both from last year). In NIH funding, the region is fourth (5,180 awards totaling $3.13 billion).
3. BioHealth Capital Region (Maryland, Virginia, and Washington, D.C.)

The BHCR takes in Virginia and Maryland, both of which benefited over the past year from the domestic “reshoring” of biomanufacturing by pharma giants. AstraZeneca in November announced $2 billion in plans for Maryland that include a major expansion of its biologics manufacturing facility in Frederick, MD, and a new clinical manufacturing facility in Gaithersburg, MD. A month earlier, AstraZeneca expanded the scope of its new manufacturing facility in Rivanna Futures, near Charlottesville, VA, into a $4.5-billion project designed to support manufacturing for weight management, metabolic, and cancer technologies, including antibody-drug conjugates. The project is expected to create 600 permanent jobs. Also last fall, Merck & Co. broke ground on a $3 billion, 400,000-square-foot Center of Excellence for Pharmaceutical Manufacturing at its longstanding site in Elkton, VA, while Eli Lilly announced plans for a $5-billion manufacturing facility just west of Richmond, VA, in Goochland County that will be the company’s first-ever dedicated, fully integrated active pharmaceutical ingredient (API) and drug product facility for its bioconjugate platform and monoclonal antibody portfolio. However, a longtime strength of the region—the headquarters presence of the NIH and FDA—is now among its most serious challenges as government funding cuts chopped the workforces of both agencies last year by 3,500 and 1,200 jobs, respectively, though the FDA in recent months has worked to hire 1,000+ new staffers to fill reviewer, inspector, and investigator roles. And in May, Gilead Sciences disclosed plans to lay off 84 employees in Rockville, MD (and 108 in Redwood City, CA) following its $7.8-billion acquisition of Arcellx.
The BioHealth Capital Region fulfills its top-three cluster ambitions by continuing to lead the nation in patents (80,808 families) while placing third in NIH funding (4,665 awards totaling $3.474 billion) and lab space (37.208 million square feet according to JLL data cited by BHCR, including 9.2 million square feet of NIH labs in Bethesda, MD). The region is fourth in jobs (135,298, according to JLL and state data cited by BHCR), but seventh in venture capital ($1.117 billion in 2025, zero in Q1 2026, according to BHCR data).
4. New York/New Jersey

The Big Apple will soon see a big biotech campus emerge, the $1.6 billion, 2-million-plus-square-foot Science Park and Research Campus (SPARC) Kips Bay, projected to create more than 15,000 jobs by combining life-science space with academic and public health facilities. Exterior demolition is scheduled for the third quarter, followed by construction next year. However, Johnson & Johnson has shifted operations of its JLABS@NYC incubator to site owner New York Genome Center, part of a corporate cutback of its incubator network. The 17-member Emerging Technology Advisory Board appointed by New York Gov. Kathy Hochul (D), who is seeking re-election this year, proposed numerous efforts in December to expand life sciences activity statewide, including a $65-million “Excellence” fund and a $40-million pre-commercialization fund. At deadline, the fate of those efforts was unknown despite a tentative agreement on May 7 of a $268-billion state budget.
In New Jersey, New Brunswick’s Planning Board in February approved the $468-million H-3, the third phase of the HELIX downtown campus, a 40-story, 554,000-square-foot tower, for which the city council approved a 30-year PILOT agreement that will generate $1.8 million a year in annual payments in lieu of taxes. In suburban Westchester County, Regeneron Pharmaceuticals is completing a $1.8-billion HQ expansion in Tarrytown but has scuttled earlier plans to expand across the Hudson River into the Rockland County village of Suffern, where the company spent $39 million to buy an old Avon Cosmetics warehouse for conversion into an infectious disease lab and a cold storage facility. In February, Regeneron hired JLL to market the site for sublease.
New York and its northern New Jersey suburbs lead the nation in NIH funding (7,033 awards totaling $4.396 billion) and are third in jobs (147,900, according to Cushman & Wakefield). From there, the region falls to the middle of the pack, placing fifth in VC ($1 billion in 2025 and about $400 million in Q1 2026, both according to PitchBook), and sixth in both lab space (25.5 million square feet, according to Colliers) and patents (12,523 families).
5. Greater Philadelphia

Eli Lilly made history in January by announcing Pennsylvania’s largest-ever biotech project, a $3.5-billion biomanufacturing site planned for Upper Macungie Township, an hour’s drive northwest of Philadelphia. Lilly plans to base 850 jobs at the plant, which will produce retatrutide and other weight loss drugs when it becomes operational in 2031. Lilly also has plans for the City of Brotherly Love, namely a 44,000-square-foot Lilly Gateway Labs innovation hub in Center City West at 2300 Market set to open later this year. And, in Philadelphia’s Old City, Thermo Fisher Scientific last November opened its East Coast Advanced Therapies Collaboration Center (ATxCC) within the BioLabs for Advanced Therapeutics incubator.

The region’s rich biotech history includes the first gene therapy Luxturna® marketed by Roche-owned Spark Therapeutics—which is completing its $575 million Gene Therapy Innovation Center in University City despite laying off more than half of its Philly staff last year. In March, TerraPower Isotopes announced plans for a $450-million radioisotope manufacturing facility designed to produce actinium-225 for cancer treatments. The project will employ 225, receive $10 million in state grants, and rise within The Bellwether District, the 1,300-acre former Philadelphia Energy Solutions refinery site. Greater Philadelphia has long benefited from innovations from its institutions, two of which won more than $100 million in NIH funding during the 2025 federal fiscal year, the Perelman School of Medicine at the University of Pennsylvania to Children’s Hospital of Philadelphia (CHOP)—which last year treated KJ Muldoon (“Baby KJ”), the world’s first patient to receive a personalized CRISPR gene-editing therapy (for CPS1 deficiency). The region’s needs for more C-suite talent and venture capital remain persistent challenges to cluster growth, stakeholders told The Philadelphia Inquirer in December, though Audrey Greenberg, chair of corporate development and “Mayo Venture Partner” at Mayo Clinic and founder of AG Capital Advisors, told the Inquirer: “I’m going to be starting my companies all here in Philadelphia, because that’s where I am.”
Greater Philadelphia improved the most this year, climbing two positions in this year’s A-List after remaining fifth in patents (17,090 families) and rising to fifth in lab space (25.9 million square feet, according to Colliers’ data cited by Pennsylvania’s Department of Economic Development or DECD) and NIH funding (3,201 awards totaling $1.94 billion). The region jumped four spots to fifth in VC ($1.31 billion in 2025, $616 million in Q1 2026, says Colliers), but dipped to seventh in jobs (88,000, also according to DECD), including nearly 10,000 with cell and gene therapy expertise.
6. San Diego

The Biotechnology Innovation Organization (BIO) expects to draw 20,000 to its BIO International Convention when it returns this month to the San Diego Convention Center. The region remains a vibrant life-sciences cluster: Novartis broke ground in February on a $1.1-billion, 466,000-square-foot global Biomedical Research center in San Diego, expected to house 1,000 employees when operational in 2029, three months after opening a radioligand therapy manufacturing facility for cancer treatments in Carlsbad, CA. Eli Lilly in March completed its $1.2-billion acquisition of home-grown Ventyx Biosciences—months after the pharma opened a Lilly Gateway Labs innovation hub with Alexandria Real Estate Equities in Torrey Pines. The J. Craig Venter Institute—whose founder died April 29 at age 79—plans this summer to move its West Coast headquarters from the University of California San Diego campus in La Jolla to the downtown Research and Development District (RaDD), a $1.6-billion, 1.7-million-square-foot campus on the city’s Pacific coastline completed last year by San Diego-based developer IQHQ—which is fighting an investor’s fraud allegations related to a $50-million investment in 2020. Home-grown F5 Therapeutics (up to 10 employees) folded in March, while two other San Diego biotechs laid off employees this year: Gossamer Bio (65 employees, nearly half its workforce, as of May 15, following a Phase III trial failure) and BioAlta (70% of its staff, which was 41 as of December 31, 2025). In February, San Diego drug developer Iambic Therapeutics inked an up-to-$1.7-billion collaboration with Takeda Pharmaceutical, which will use Iambic’s AI technologies and wet lab capabilities to design and develop small molecule drugs. And global contract development and manufacturing organization (CDMO) Bora Biologics, in January, opened a $30-million expanded manufacturing facility with two to four 2,000-liter bioreactors, corresponding seed trains, and advanced downstream processing equipment.
“America’s Finest City” and vicinity stayed third in VC ($1.9 billion in 2025, says PitchBook, $743 million in Q1 2026 according to a GEN spot-check of recent deals) and fourth in patents (18,314 families) but dipped to fifth in lab space (28.685 million square feet, according to CBRE). While the San Diego region last year rose to ninth in NIH funding (2,001 awards totaling $1.357 billion), it slid to ninth in jobs (71,448, according to year-old BIOCOM California data).
7. North Carolina

Always strong on drug manufacturing, North Carolina is among the biggest beneficiaries of biopharma’s reshoring push. In April, AbbVie announced a $1.4-billion, 185-acre drug production facility in Durham County near Research Triangle Park (RTP), expected to employ 734. Roche’s Genentech subsidiary in January expanded to $2 billion its planned investment in its first East Coast manufacturing facility in Holly Springs, NC, which broke ground last year and is set to support 500+ manufacturing jobs when operational by 2029. And in November 2025, Novartis said it will expand Tar Heel State operations into a flagship manufacturing hub by adding capabilities for sterile filling of biologics into syringes and vials at its current Durham site, constructing two new Durham facilities for manufacturing biologics and sterile packaging, and building a new Morrisville, NC, site to produce solid dosage tablets and capsules, including packaging. Morrisville is where Novartis also plans to build a 56,200-square-foot facility focused on API manufacturing for solid dosage tablets, capsules, and RNA therapeutics, a project announced April 30. Manufacturing sites account for most of the combined $24.5 billion in new or expanded facilities with a potential 15,000+ new jobs that life sciences companies have announced statewide since 2021, according to the state-funded North Carolina Biotechnology Center. As for startups, Raleigh-based Slate Medicines launched in February with $130 million in Series A financing to fund development of therapies led by its migraine candidate, the anti-PACAP monoclonal antibody SLTE-1009 licensed from Zhongshan, China-based DartsBio Pharmaceuticals, and set to start Phase I trials in mid-2026.
The Tar Heel State climbed to fourth in VC ($1.6 billion in 2025, $276.8 million in Q1 2026, both according to the state-funded North Carolina Biotechnology Center). But North Carolina showed consistency on the other criteria, ranking seventh in NIH funding (2,248 awards totaling $1.589 billion) and lab space (18.6 million square feet, according to JLL), and eighth in jobs (76,000, says the Center) and patents (5,992 families).
8. Los Angeles / Orange County, CA

The region’s biopharma anchor Amgen broke ground last fall on a $600-million center for science and innovation being built within its Thousand Oaks, CA, headquarters campus, set to integrate research & development and process development teams to smooth the transition from drug discovery to commercial manufacturing. “With the first shovel in the ground, we’re reaffirming something essential: We discover here, we manufacture here, we deliver for patients from Thousand Oaks to all around the world,” Amgen chairman and CEO Robert A. Bradway said. Regional industry group BioscienceLA CEO Stephanie Hsieh recently acknowledged the region’s fragmentation as a challenge—from 88 cities in LA County alone, to the numerous county, city, and private agencies focused on growing the bioindustry— while citing strengths such as corporate anchors Amgen, Takeda Pharmaceutical, and Gilead Sciences-owned Kite Pharma, plus institutions like USC, UCLA, Cedars-Sinai, and City of Hope.
California signaled interest in growing the region’s biopharma industry last August when the state-funded California Jobs First Regional Investment Initiative awarded $23.92 million to a coalition led by Los Angeles County’s Department of Economic Opportunity (DEO) toward four programs intended to create 10,000 jobs by 2030. Most of the money ($19 million) was approved for a DEO revolving loan fund to support startups, especially those looking to graduate from the Larta Institute’s commercialization and capital access accelerator into lab space within Los Angeles County. Larta was awarded $3.3 million to expand its Heal.LA Bioscience & Healthcare Accelerator and assist small startups via its Larta Impact Fund, a revolving loan fund.
Los Angeles/ Orange County would still lead the nation in jobs, based on a year-old BIOCOM California tally of 155,571, which also includes San Bernardino and Ventura counties; figures run as low as 116,000, compiled last year for the four counties plus Riverside and Santa Barbara counties (regional industry group SoCalBio). The region finished seventh in patents (7,211 families), eighth in lab space (11.7 million square feet, according to JLL), and 10th in both NIH funding (1,911 awards totaling $1.243 billion) and VC ($500 million in 2025, zero in Q1 2026, according to PitchBook).
9. Chicagoland

At least one developer has pivoted to a large non-biotech tenant to help fill a Chicago campus once envisioned as a life-sciences mecca: Trammell Crow in March inked a $100-million, 169,860-square-foot lease with candy/chocolate giant Mars to base 600 jobs at 400 North Aberdeen Street within the Fulton Market campus. Other biotech spaces are in the works: In North Chicago, Rosalind Franklin University of Medicine and Science plans to nearly double the size of its Helix 51 biomedical incubator to just under 13,000 square feet by adding 6,000 square feet of new lab and office space, citing growing demand from early-stage biotechs. The expansion is expected to create space for up to 10 additional companies. Also in North Chicago, home-grown AbbVie announced plans to build two new API manufacturing facilities totaling $380 million at its campus in the Chicago suburb. The facilities—designed to support production of next-generation neuroscience and obesity treatments—are set to be fully operational in 2029. However, AbbVie opted to build its planned $1.4-billion biomanufacturing campus not in North Chicago but 821 miles southeast in Durham, NC. Across Illinois, biotech stakeholders have applauded Gov. J.B. Pritzker (D) for proposing to sweeten the state’s Research & Development Tax Credit program by allowing companies to transfer their credits for cash. “This is a transformative step for our startup and growth-stage ecosystem,” stated John Conrad, president and CEO of the Illinois Biotechnology Innovation Organization (iBIO). Pritzker is seeking a third term in November vs. Darren Bailey (R).
The Windy City and vicinity rank sixth in both NIH funding (2,658 awards totaling $1.607 billion) and jobs (94,000, according to statewide industry group Illinois Biotechnology Innovation Organization or iBIO). The region places ninth in patents (5,569 families) and VC ($917.677 million in 2025, says iBIO, zero in Q1 2026,
10. Seattle

Seattle and the Greater Puget Sound’s strong base of academic and other nonprofit research institutions helped the region achieve consecutive years of Nobel laureates: Mary E. Brunkow, PhD, of the Institute for Systems Biology in Seattle co-won the 2025 prize in Physiology or Medicine a year after David Baker, PhD, director of the Institute for Protein Design at University of Washington (UW), co-won the 2024 prize in Chemistry. A UW spinout, Seattle-based 3D tissue model developer Curi Bio, closed in December on a $10-million Series B financing led by South Korean contract research organization DreamCIS. In April, Achieve Life Sciences (based in Seattle and Vancouver, BC) announced an up-to-$354 million private placement whose purposes include funding a Phase III trial and future commercialization of e-cigarette cessation candidate cytisinicline, while Athira Pharma landed up to $236 million in conjunction with acquiring exclusive rights from Sermonix Pharmaceuticals to the Phase III metastatic breast cancer candidate lasofoxifene. AGC Biologics, a global CDMO, expanded its regional research footprint last fall by signing a 37,575-square-foot lease at Element Research Center in Bothell, WA. However, Astellas Pharma told Washington state officials in April it will shutter the Seattle site of its Universal Cells subsidiary by 2028, with 50 employees to be impacted via layoffs or transfers to South San Francisco, CA, or Westborough, MA.
Seattle and its suburbs placed highest at eighth in both NIH funding (eighth with 1,892 awards totaling $1.572 billion) and VC ($1.06 billion in 2025, zero in Q1 2026, according to industry group Life Science Washington). The region was ninth in lab space (11.46 million square feet, according to regional real estate firm Flinn Ferguson Cresa) and 10th in both jobs (48,765 according to Life Science Washington) and patents (5,416 families).
The post Top 10 U.S. Biopharma Clusters 2026 appeared first on GEN – Genetic Engineering and Biotechnology News.



 portfolio brand, including the Tocris
portfolio brand, including the Tocris